
Software consultant
Mountain View, CA 94040
pcm@rahul.net
http://www.rahul.net
This is a draft paper for a poster session at the
Sixth
Foresight Conference on Molecular Nanotechnology
This paper describes software under development that should make it easy to specify complex artificial molecules by simple scripts which describe moieties and how they attach to each other.
Each moiety description includes a list of dangling bonds plus a dihedral angle to specify the orientation of each dangling bond. The relative positions of the atoms in the moiety are specified by the bonds between them and as many dihedral angles as are needed to make it unambiguous.
The scripts are written in Python and use objects from Konrad Hinsen's Molecular Modelling Toolkit, which produces a powerful and flexible language without making the scripts hard to read or reinventing much syntax.
Some people who think about nanotech design software focus on visual ways of specifying molecular systems. I doubt that a primarily graphical approach will scale up adequately. Small proteins or Drexler's example parts appear to be stretching the limits of what I can comprehend via a single picture. There are severe limits to how accurately I can position objects using a mouse when trying to control 6 degrees of freedom. GUI's impose important limitations on parallel operations that we might want to do on many disparate parts of a system.
The specific problem that provoked me into starting this software was the desire to find a good molecular equivalent to Lego (tm) Blocks.
I chose MMTK as the starting point for my efforts because it had the best combination of general purpose molecular modelling code, readability, and flexibility. Python's ability to import and parse code at runtime is an added bonus, which avoids the need to invent yet another scripting language.
The main drawback of Python is inadequate provision for type checking, which can lead to confusion and errors in passing objects to imperfectly documented parts of the system.
Unlike most software tools where there is a clear dividing line between the user interface and the internal implementation, the use of Python and MMTK provides a high-level interface designed specifically for most end users, but also allows sophisticated users access to most low-level methods in the system.
Users will need to learn some of the Python programming language, but it is unlikely that they will need to learn more than 5% of what it would take to become a serious Python programmer.
I start by creating the following straightish 4 carbon moiety:
Detailed Example
This example shows how my search for a molecular Lego (tm) block has
been proceeding.
name = 'diamond4carbon'
C1 = Atom('C')
C2 = Atom('C')
C3 = Atom('C')
C4 = Atom('C')
b12 = Bond(C1,C2)
b23 = Bond(C2,C3)
b34 = Bond(C3,C4)
da1 = Dihedral(C1,C2,C3,C4,180*deg)
amber_atom_type = {C1: 'CT', C2: 'CT', C3: 'CT', C4: 'CT' }
dC1a = DanglingBond(C1, Dihedral(None,C1,C2,C3,60*deg))
dC1b = DanglingBond(C1, Dihedral(None,C1,C2,C3,-60*deg))
dC1c = DanglingBond(C1, Dihedral(C3,C2,C1,None,180*deg))
dC2a = DanglingBond(C2, Dihedral(None,C2,C3,C4,120*deg))
dC2b = DanglingBond(C2, Dihedral(None,C2,C3,C4,-120*deg))
dC3a = DanglingBond(C3, Dihedral(C1,C2,C3,None,60*deg))
dC3b = DanglingBond(C3, Dihedral(None,C3,C2,C1,120*deg))
dC4a = DanglingBond(C4, Dihedral(C2,C3,C4,None,60*deg))
dC4b = DanglingBond(C4, Dihedral(C2,C3,C4,None,-60*deg))
dC4c = DanglingBond(C4, Dihedral(C2,C3,C4,None,180*deg))
hooknorth = Hook(None, [dC1c])
hooksouth = Hook(None, [dC4c])
hookup = Hook(None, [dC2b, dC4b])
hookdown = Hook(None, [dC1a, dC3b])
hookeast = Hook(None, [dC2a, dC4a])
hookwest = Hook(None, [dC1b, dC3a])
Most of the remaining code in this example comes from a script named
lego6.py, and
roughly corresponds to the chronological steps by which I proceeded.
Each excerpt is followed by some explanations.
ff = GenericForceField('mm3')
world = InfiniteUniverse(ff)
block1 = Group('diamondoid_fragment')
join = Joiner(block1)
width = 5
join.repeatGroup('hookeast', 'hookwest', width)
join.repeatGroup('hooksouth', 'hooknorth', 5, 1, [180*Units.deg]*width)
join.repeatGroup('hookup', 'hookdown', 2)
result = join.getResult()
result.setPositions(ff)
The name result is now bound to a Group that is a roughly
rectangular slab of 5 by (4*5)=20 by 2 carbons. The 1 in the
middle repeatGroup call is an optional argument that controls
whether the repeated block is reused or cloned. The list of width
angles after it specifies the angles for the dihedrals about the single bond
that connects each hooksouth Hook to each hooknorth.
Ideally the software should be able to deduce that those are the only
reasonable dihedral angles for those bonds, but it will take more programming
effort to find a way of doing this that scales up well.
The other hooks in this example all involve more than one bond, which
automatically constrains the dihedral in a way that the software can
deduce if the constraints are consistent with each other.
a_hole = result.findAtomByName('diamond4carbon.diamond4carbon7.C4')
surface_point = Positioner.findNearestPointOnSurface(result, a_hole)
surface_vector = surface_point - a_hole.position()
dist_to_surface = surface_vector.length()
surface_dir = surface_vector.normal()
cylinder1 = Cylinder(a_hole.position() - 0.1*Units.Ang*surface_dir,\
2.5*Units.Ang, (1*Units.Ang+dist_to_surface)*surface_dir)
I want to dig a hole in one face of the slab. I looked at what the script
had produced so far (using a version of RasMol that I
have begun to integrate into MMTK), decided on an atom to center the hole
around, and got it's name by clicking on it. I then created a line from
that atom through the nearest point on the convex hull of the system (a set
of triangles bounding the atom positions as closely as possible without
making any concavities), and generated a cylinder about that line (extending
the line a bit to make sure it included that atom and any other that might
be on the cylinder boundary.
atoms_in_cyl1 = result.atomsWithinRegion(cylinder1) result.removeAtoms(atoms_in_cyl1, None)This removes the atoms enclosed by the cylinder.
c_below = result.findAtomByName('diamondoid_fragment.diamondoid_fragment32.C3')
nitrogen = result.generateNewAtom('N')
result.replaceAtoms([c_below], [nitrogen])
result.fillDanglingBonds(ff, 'N', 8, 3)
I saw that the carbon atom remaining at the center of the hole's bottom had a
dangling bond remaining that might interfere with the way I hoped to insert
things into the hole, so I replaced it with a nitrogen.Six places where carbons had been removed left places to which 3 dangling bonds were pointing; the fillDanglingBonds call finds all such positions and fills them with atoms of symbol 'N', MM3 type 8.
def filter2n(atom): # define a function which returns true
if atom.symbol != 'C': # for carbons bonded to exactly 2 nitrogens
return 0
bonded_to = atom.bondedTo()
countn = 0
for a in bonded_to:
if a.symbol == 'N':
countn = countn + 1
return countn == 2
carbons_bonded_to_2_ns = filter(filter2n , result.atomList())
selected_ns = []
replace_cs = []
for a1 in carbons_bonded_to_2_ns:
for a2 in a1.bondedTo():
if a2.symbol == 'C':
c = a2
a1_pos = a1.position()
for a2 in a1.bondedTo():
if a2.symbol == 'N':
x_product = (a1_pos - c.position()).cross(a2.position() - a1_pos)
if surface_dir * x_product > 0: # select by orientation
selected_ns.append(a2)
replace_cs.append(result.generateNewAtom('C'))
result.replaceAtoms(selected_ns, replace_cs, ff)
for i in range(len(carbons_bonded_to_2_ns)): # make double bonds:
result.attachBondBetween(carbons_bonded_to_2_ns[i], replace_cs[i])
I want to create some double bonds along the edge of the hole to make
it moderately stable by itself, but still able (I hope) to react with
similar double bonds on the knob I plan to insert into the hole.
First I search for all the carbon atoms attached to two nitrogens, which gives me three carbons associated with the six nitrogens I added in the prior step. I then identify three of the nitrogens on the basis of an arbitrarily chosen orientation from the selected carbons, replace those three nitrogens with carbons, and create double bonds from the previously selected carbons to the new carbons. The double bonds will be somewhat strained, but probably no more so than in figures 8.9 and 8.10 in NanoSystems (cubene and adamantene).
I'm now satisfied with the hole, and turn my attention to adding a knob on the other side of the slab that will fit into such a hole on another "Lego block". I create the following file cyanide:
name = 'Cyanide'
C = Atom('C')
N = Atom('N')
b = Bond(C, N, 3)
mm3_atom_type = { C : 4, N : 10 }
db = DanglingBond(C, Dihedral(None, N, C, None, 0.0))
hook1 = Hook(None, [db])
Back to the main script:
hook3_list = []
for db in result.danglingbonds:
if nitrogen in db.a1.bondedTo():
hook3_list.append(db)
hook3 = Hook((result, hook3_list))
join2 = Joiner(result)
layer = Group('lego4')
join2.joinGroups(hook3, layer.hook1, 0)
result = join2.getResult(0)
This creates a knob of three nearby cyanides. (Possibly the nitrogens will
bond with neighboring cyanide carbons to form a ring. That would probably be
equally close to the shape and reactivity I want).
result.setPositions(ff) result.hydrogenateDanglingBonds(ff)This defines positions for the atoms in the knob, and adds fills all remaining dangling bonds with hydrogens.
result2 = result.clone()
result2.name = 'd42'
face2 = []
for a in result2.atomList():
if a.symbol == 'N':
bonded = a.bondedTo()
if len(bonded) == 1 and bonded[0].symbol == 'C':
face2.append(a)
face2 = [face2[0], face2[2], face2[1]]
PositionAdjacent(result,carbons_bonded_to_2_ns, result2,face2, [2*Units.Ang]*3)
Finally, I duplicate the molecule, pick three atoms on the knob of the
new molecule, and rotate/translate it so those three atoms are 0.2 nanometers
(2 angstroms) from three of the carbons on the edge of the original molecule's
hole (actually, it can't satisfy those three constraints, and the actual
distances are 0.2 to 0.32 nanometers (2 to 3.2 angstroms); see the
PositionAdjacent description
for details).
This produces a pair of molecules with potentially reactive double and triple bonds positioned 0.3 to 0.4 nanometers (3 to 4 angstroms) apart.
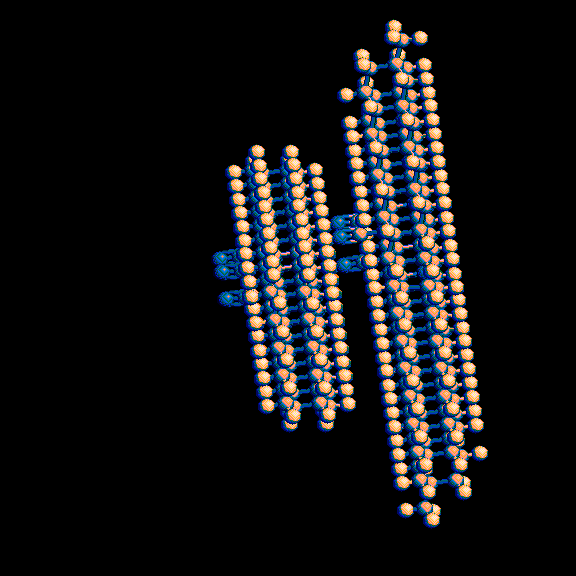

3-D representations of these images are available at
http://www.rahul.net/pcm/mmtk/mnt6_images.html.
I plan to add another hole and another knob at the other end of the slab and run molecular dynamics simulations on the result to determine whether the three double bonds / triple bond pairs react to bond the two molecules together in a reasonably rigid result, and if so how closely they have to be positioned before they spontaneously form these bonds in a predictable one of the three possible orientations (each 120 degrees apart). If that works, I will tweak the shapes a bit so they fit well when I put many of them together into larger structures.
I haven't yet addressed constructing systems which are capable of motion.
I started with diamondoid systems because they are simpler to model, but
I am equally interested in solution-based systems.
Will this approach work for floppy molecules such as DNA or proteins?
I expect some problems associated with handling under-constrained dihedrals.
I don't want to tackle the protein folding problem, but I also want to
minimize the need for specifying dihedrals by users who are constructing
systems from standardized components.
When I have the diamondoid tests sufficiently debugged, I will try designing
some building blocks using DNA that have some hope of producing controlled
motion and of being produced with today's technology.
At very least I will need to supplement this with the ability to import
PDB files as rigid components. I will also try to find reasonable ways
to relax the rigidity requirement without allowing the software to guess
at absurd conformations.
I may try include using distances between atoms to specify constraints.
Also, the toolkit is still in it's infancy as this paper is being written;
this version is designed more to provide a taste of what the system can
accomplish than as a programming guide.
Updated information will be available at
http://www.rahul.net/pcm/mmtk/,
along with more example scripts and pdb files they produce
(such as "Hello, world!" written in hydroxyls on diamondoid),
and source code.
This is where a majority of the construction work is done. It goes through
each subgroup, assigning positions to each atom to meet the bond and dihedral
constraints within each subgroup, and then positions the subgroups to satisfy
constraints on bonds between subgroups.
Within the base level subgroups, it starts by setting the first bond to
an arbitrary position, then keeps looking through the list of bonds for
those which have one atom positioned and setting the position of the other
atom. When it notices that it's positioning has caused a dihedral
constraint to dictate the position of a neighboring bond, it gives priority
to satisfying that constraint. When it notices that both atoms in a bond
which it hasn't processed have already been positioned, it deduces that
it has found a cycle, and rotates any under-constrained dihedrals along
that cycle as needed to make the bond under consideration the right length.
If rotating dihedrals doesn't produce a good enough result, it sometimes
tries bending some bond angles.
Arbitrary lines can be drawn using AddPolygon and AddPolyLines, followed
by RefreshScreen() and Interact().
The three positions on the first
object are used to specify two triangles at the specified distances above
and below the plane of the three atoms. It chooses the triangle farther
from the first object's center of mass (a crude way of avoiding putting atoms
on top of each other; I expect to replace this with something based on a
Delaunay triangulation to maximize the distance from the surface).
It then positions the second object so the three atoms from it are in the
plane of the chosen triangle. The first of those atoms is aligned over the
corresponding atom in the first object, the second is positioned in the same
direction as the second atom in object 1, and the third is placed on the
same side of the line between atoms 1 and 2 as the third atom in object1.
(This algorithm will probably be replaced with one that spreads the error
equally between pairs of atoms).
The atom from the second object is positioned so as to maximize it's distance
from the atoms that are neighbors of the specified atom in object 1 (neighbors
being defined by the Delaunay triangulation).
Then the second object is rotated so that the atom specified on object 1
meets the equivalent condition for the atoms in object 2.
This is designed to work with atoms on the convex hulls of the objects,
but will break down as that assumption gets further from the truth.
I plan to try approaches such as used in DelaunayTriangulation.removeFacesBiggerThan
to handle this in a way more like what molecular docking software does.
Prospects
If such building blocks could be created before a real assembler exists
(a big assumption which I know I can't justify), then a positioning device
with a fair amount of error might be able to construct a wide variety of
atomically precise rigid structures.
Future Software Enhancements
While I believe my initial approach of relying on dihedrals to specify
shapes provides a powerful method of indicating which parts of the
moiety need to be constrained without constraining things which should
adapt to the conditions under which it is being used, it has proven harder
that I expected to use them to design good components.
The existing replaceAtoms function, combined with simple pieces of Python
code, provides some of what I want in a search and replace function, but
only works on single atoms so far, and the search part is an accidental
byproduct of the power created by MMTK's approach. I plan to investigate
whether I can produce a regular expression language that improves on this
search approach.
I plan to support using a wide variety of geometric objects, including
unions and intersections of geometric objects (constructive solid geometry
as used in CAD software), plus the ability to fill regions defined by
molecular shapes read from PDB files with user-defined patterns of atoms.
Currently the joining and positioning steps in the example above
take about 150 seconds on a 300 mHz Pentium II, and appear to scale up
roughly in proportion to the square of the number of atoms. I plan to
identify the bottlenecks and replace them with O(n) or O(n log n) algorithms
where possible.
Currently I use the system as a compiler, rebuilding most moieties
from scratch each time I test something. It should be possible to use
it more like an interpreter (saving intermediate steps) with many objects
an operations on them selected via a GUI (presumably derived from RasMol)
whenever that can be made quicker than typing the text-based methods.
I have been trying to minimize use of absolute reference frame in favor of
relative positions, partly in hopes of being close to how an actual assembler
would operate, but mainly because the setPositions method includes
some random decisions which ensure that no 2 runs produce the same atom
positions. I will try to find out why seeding the random number generator
doesn't make the arbitrary choices repeatable, as that will probably make
some geometric operations easier.
Warning: Nearest available angle 48.24 degrees from expected
Dihedral(Atom Lego3.C1,Atom Lego3.N1,Atom
Lego3.C2,None,-60.0*deg); expected -60.00
Traceback (innermost last):
File "Programs/lego1.py", line 32, in ?
result.positionDanglingBonds(ff)
File "./CompositeObject.py", line 1772, in positionDanglingBonds
(dir, constraint, cmbond, far_bond) =\
File "./CompositeObject.py", line 985, in nextBondDirection
raise ConformationError
ConformationError
where what is really needed is a way to identify what constraint was
inconsistent with the constraint it failed to satisfy here.
Reference Manual
There is no well-defined line between what methods are intended for users
writing scripts that construct systems, and methods that are intended for
programmers implementing more methods, but this section will try to give
specifications of those methods that are most useful for script writers.
Classes/Methods
Notations used:
(ChemicalObject is a subclass of GroupOfAtoms)
Creates a new Group by parsing a file in the Groups directory.
This class creates Bond's to attach DanglingBond's from two or more
Group's. It is intended to be used before atom positions have been
defined, and doesn't assign positions.
Returns a Group or a Molecule depending on whether there are any
DanglingBond's remaining.
This class is the beginning of a Python interface to
RasMol.
Creating an instance of RasMol for an object and then calling Interact()
should be almost equivalent to writing the object to a PDB file, and then
viewing it with the standard RasMol program. The main difference is that
atom labels will be the MMTK names, which are often too long to fit in the
PDB format. Also, dangling bonds will be displayed and labelled if
positionDanglingBonds and labelDanglingBonds have been called.
Built on top of Ken Clarkson's
Hull 1.0 code, this class defines the convex hull of the atom positions,
and the Delaunay triangulation (tetrahedra bounded by the shortest lines
between atoms).
This is intended to position two completed molecules in a good position
with their surfaces near each other. Each atom in the list of atoms
associated with the first object is positioned as close as feasible to
the distance with the same index in the distances list from the atom
with the same index in the list of atoms from the second object.
Two cases are supported so far:
Returns the point on the convex hull closest to the Atom's position.
May return None, a Vector or pair of Vector's (points), or a
GeometricalObject3D. Implemented for:
Provides a set of points suitable for displaying an outline (sometimes quite
crude) of the object in a 3-d GUI (RasMol, GnuPlot, etc.).
Like pointNearest, but returns None if result isn't inside Polygon.
Prism must be constructed from a convex Polygon which is a sequence of
coplanar points, ordered clockwise as seen from inside prism.
A infinite line.
References
Lego is a trademark of the LEGO Group.
Corresponding Address:
Peter McCluskey
1571 El Camino Real W.,#15
Mountain View, CA 94040
(650) 967-3359
pcm@rahul.net
http://www.rahul.net/pcm
Foresight materials on the Web
are ©1986-1998 Foresight Institute. All rights reserved.
Last updated 19Oct98. The URL of this document is not yet determined;
see the following for more information about it:
http://www.rahul.net/pcm/mmtk/
http://www.foresight.org/Conferences/MNT6/
http://starship.skyport.net/crew/hinsen/mmtk.html